

Next: Transition matrices
Up: Nucleotide substitution models
Previous: Nucleotide substitution models
Contents
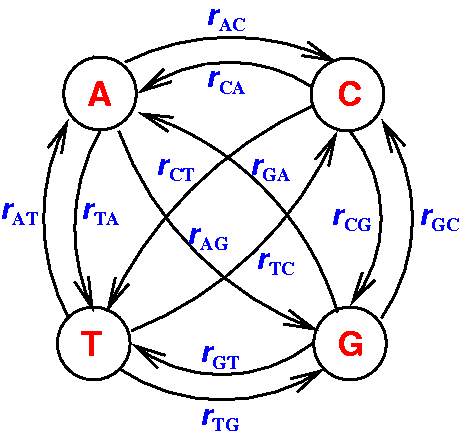
Replacements within DNA sequences can be described and modelled by a Markov
process with four states. Each state represents one base --
Adenine, Cytosine,
Guanine or Thymine (see figure 2.2).
Figure 2.2:
Markov model for nucleotide evolution in DNA sequences
|
|
Lots of assumptions are made in order to make phylogenetic reconstructions more
computationally feasible. First, each nucleotide is supposed to evolve
independently of other sites evolution and of its past history. We suppose
there is no interaction between sites and we treat them independently. Further, the
Markov process of substitution is assumed to be the same across all sites
(spatial homogeneity). Finally, the process is assumed to remain
constant over time (stationary) and time homogeneous, i.e.,
nucleotide frequencies and substitution rates can be assumed constant
through time and across all sites in an alignment.
One might concede that assumptions made for the nucleotide evolutionary
process are not strictly valid. Actual data shows some discrepancies, e.g.,
heterogeneous selection pressure, unequal base frequencies among species, ....
We can relax these assumptions and allow for substitution rate variation
across sites with the gamma model of Yang (1994).
It is also possible to use multiple substitution processes simultaneously when
heterogeneous data are analysed (see section 2.4.2).
In spite of their name, DNA models can naturally be used for the
treatment of the loops within RNA sequences (see figure 2.3).
In RNA loops, nucleotides are not subject
to any structural constraints and they are assumed to evolve
independently from other sites. Therefore, the use of similar
Markov models for nucleotide evolution in RNA loops is appropriate.
Next: Transition matrices
Up: Nucleotide substitution models
Previous: Nucleotide substitution models
Contents
Gowri-Shankar Vivek
2003-04-24